Alimentation, santé globale Temps de lecture 6 min
CroCoDeEL : un outil puissant de détection des contaminations croisées entre échantillons
CroCoDeEL, un outil puissant d'aide à la décision permettant de détecter et de quantifier la contamination croisée entre échantillons lors d'une analyse métagénomique. Il s’appuie sur la modélisation linéaire et un classificateur supervisé pré-entraîné et identifie des schémas de contamination spécifiques dans les profils d'abondance des espèces.
Publié le 13 juillet 2026
Le séquençage métagénomique permet de caractériser à haut débit des communautés microbiennes complexes telles que le microbiote intestinal humain. Cette approche demeure toutefois exposée à un biais technique majeur : la contamination croisée, un transfert accidentel d’ADN d’un échantillon à un autre lors des étapes de préparation en laboratoire.
Non détectée, cette contamination conduit à attribuer à un échantillon des micro-organismes en réalité absents dans celui-ci, compromettant la validité des résultats.
CroCoDeEL :
Détecter et Quantifier
les Contaminations
Pour y remédier, des chercheurs d'INRAE (unités MetaGenoPolis et MaIAGE) et de l'IRD ont développé CroCoDeEL (CroCoDeEL est disponible librement sur GitHub), un logiciel dédié à la détection et à la quantification de telles contaminations.
L'outil s'appuie sur l'analyse des profils d'abondance taxonomique afin d'identifier des signatures de transfert d’espèces entre échantillons, signe de contamination. Grâce à des approches combinant modélisation statistique et apprentissage automatique, CroCoDeEL permet d'en estimer le niveau (y compris à très faibles taux) et d’identifier l’échantillon source.
Publiée dans Nature Communications, en mai 2026, sous le titre : CroCoDeEL: accurate control-free detection of cross-sample contamination in metagenomic data, l'étude associée révèle des contaminations critiques passées inaperçues dans plusieurs travaux de référence fortement cités : certains transferts microbiens mère-enfant décrits dans la littérature relèveraient ainsi d'artefacts techniques plutôt que de transmissions biologiques avérées, invitant à une réévaluation de résultats antérieurs.
Entretien avec l'un des auteurs de l'étude : Guillaume Gautreau, chercheur dans l'unité INRAE MaIAGE, Mathématiques et informatique appliquées du génome à l'environnement.
Pourquoi avoir voulu automatiser la détection des contaminations entre les échantillons lors des séquençages haut débit ?
Le séquençage haut débit génère un grand nombre de données à partir d'échantillons eux aussi en grand nombre. La détection des contaminations peut se faire humainement mais cela est fastidieux et relève d'un travail de titan. Avec les quantités d'échantillons analysés lors de séquençages haut débit cela devient vite ingérable et il est toujours possible de passer à côté de contaminations. Par exemple, nous travaillons sur le projet French Gut où 100 000 échantillons seront analysés. Comme la combinatoire des contaminations à envisager augmente au carré, il devient impossible d’analyser un si grand nombre d'échantillons traités sans une méthode bioinformatique dédiée.
Par ailleurs, les astuces de détection existantes reposant sur des témoins négatifs, sont insuffisamment utilisées et ne permettent pas toujours de détecter la contamination croisée au sein des échantillons non témoins. Parallèlement, les approches bioinformatiques au niveau des souches ne distinguent pas toujours une contamination réelle d’un partage naturel de souches et elles manquent de sensibilité.
C’est pour toutes ces raisons que l’automatisation de la détection des contaminations s’est rapidement imposée.
Comment expliquer les contaminations entre échantillons d'ADN ?
Le phénomène de contamination peut advenir de différentes manières lors de la manipulation des échantillons. Il faut être vigilant sur la manière dont on place les échantillons sur les plaques pour les analyses, particulièrement avec des échantillons ayant une faible biomasse (ex : méconium), sur la manière dont on fait les extractions d'ADN, notamment quand ce sont des extractions mécanisées, parce qu’il peut y avoir des projections de petites gouttelettes qui vont contaminer les échantillons qui se trouvent à côté. Il y a beaucoup de robots maintenant, cependant les robots aussi peuvent provoquer des contaminations.
De façon plus anecdotique, il existe aussi des "contaminations" bioinformatiques. Les données produites lors de séquençage haut débit représentent des volumes importants, et parfois il peut arriver qu'on mélange des fichiers sans s'en rendre compte parce qu'on a des processus automatisés. Ce mélange est généralement constaté mais il peut faire perdre du temps en faussant les résultats de la même manière qu'une contamination biologique, cela induit une contamination dite in silico.
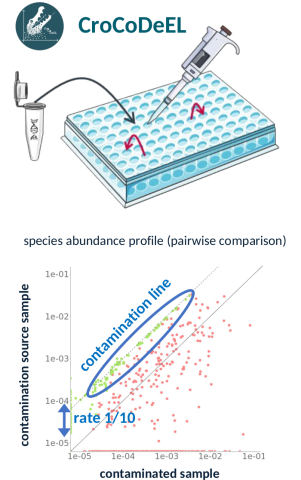
Figure : Comment CroCoDeEL repère une contamination ? Quand on manipule des dizaines d'échantillons sur plaque, un peu de matériel peut passer d'un puits à l'autre. Cela laisse une signature dans les données quand on compare deux échantillons. Les espèces amenées par la contamination, absentes de l'échantillon avant qu'elle ne survienne, s'alignent alors le long d'une même diagonale, la « droite de contamination ». Sa position indique l'ampleur du phénomène : ici, le contenu en espèces de l'échantillon contaminé provient de sa source de contamination à hauteur de 10%.
Dans votre article publié dans Nature Communications vous détaillez des analyses faites avec CroCoDeEL sur des études déjà publiées ?
Oui, nous avons appliqué le logiciel CroCoDeEL sur un nombre d'études conséquent, et on s'est rendu compte qu'il y avait des contaminations dans à peu près toutes les études, c'est donc un phénomène globalement répandu. Ces contaminations sont généralement faibles mais elles font perdre de la qualité aux résultats, et potentiellement, cela peut coûter plus cher parce qu'il faudra plus d'échantillons pour obtenir un signal sans le bruit de fond des contaminations.
Parmi ces études, il y a deux études, que nous détaillons dans l'article, qui présentent des contaminations très importantes ou bien des contaminations qui portent sur des choses assez critiques.
La première étude s’intéresse aux jumeaux au Royaume-Uni. C'est une étude qui est très citée (TwinsUK), en utilisant CroCoDeEL, on a constaté qu'il y avait 202 échantillons sur les 1004 qui étaient "hyper" contaminés. Il est clair que l'impact sur les résultats est très important, ces échantillons contaminés présentent une richesse en espèces aberrante. Nous avons donc montré que ce problème ne peut pas être mis de côté. Toutes les analyses qui sont basées sur TwinsUK sont susceptibles d'être biaisées et vont devoir être reconsidérées.
Mais l'impact est plus large que la seule étude TwinsUK. Nous avons aussi appliqué la méthode à une étude comparant les microbiotes de mères et de leurs nourrissons (Ferretti et al., 2018). Sur 182 échantillons, CroCoDeEL en a repéré 48 comme contaminés. Dans notre article, nous montrons que certaines espèces décrites comme les premiers colonisateurs passagers du bébé ont l'air d'être sur la "droite de contamination", c'est pourquoi nous pensons qu'une partie des conclusions de cette étude est biaisée. L'étude observait une diversité élevée chez les nourrissons au premier jour. Une fois les échantillons contaminés retirés, cet écart disparaît. Une partie de ce qui était pris pour un signal biologique vient donc probablement de la contamination. Signe que le problème est pris au sérieux, dans un article récemment paru dans Nature portant sur les transferts de microbiote entre bébés en pouponnière, les auteurs ont utilisé CroCoDeEL pour écarter les échantillons potentiellement contaminés (Ricci et al., 2026).
CroCoDeEL est librement accessible à toute la communauté scientifique et vous avez développé une interface d’interprétation des résultats, vous pouvez nous en dire plus ?
L'interface d'interprétation (CroCoDeEL-interpreter) permet d'explorer les résultats visuellement, directement dans le navigateur. On y retrouve les graphiques de comparaison entre échantillons, on peut y intégrer le plan de plaque et des données contextuelles. On obtient des visuels où les droites de contamination se repèrent d'un coup d'œil. L'idée, c'est que même quelqu'un qui n'est pas expert puisse comprendre pourquoi tel échantillon est signalé et juger par lui-même si la contamination est réelle et si elle est suffisamment élevée pour éliminer un échantillon (le chercheur peut parfois préférer garder un échantillon si la contamination est minime). L'interface génère aussi des rapports automatiques illustrés, qui rassemblent les cas suspects et leurs graphiques : de quoi documenter facilement le contrôle qualité d'un jeu de données, ou le partager avec des collègues.
On peut lancer directement les prédictions de CroCoDeEL depuis cette interface, sans installer quoi que ce soit. Un labo qui ne sait pas déployer un logiciel de bioinformatique n'a plus qu'à ouvrir la page pour analyser ses données. Et comme tout se passe dans le navigateur, sur la machine de l'utilisateur, les données ne sont jamais envoyées sur un serveur, un point important quand on travaille sur des données confidentielles.
Quelles sont les prochaines étapes pour CroCoDeEL ?
Deux chantiers. Le premier, systématiser le recensement des contaminations détectées par CroCoDeEL dans les études publiées et rendre cette information accessible. Quelques études sont déjà consultables dans l'onglet « Datasets » de CroCoDeEL-interpreter mais nous souhaitons aller plus loin. Ainsi, un chercheur qui réutiliserait ces données saurait d'emblée quels échantillons sont suspects.
Le second, plus ambitieux : la décontamination. Aujourd'hui, un échantillon trop contaminé est simplement écarté. Or, on jette de la donnée coûteuse à produire. Nous explorons la conception d’une méthode in silico capable de retirer le signal de contamination tout en préservant ce qui appartient vraiment à l'échantillon. Toute la difficulté est là : une espèce amenée par contamination pourrait aussi être naturellement présente. Nettoyer sans effacer ce signal-là, c'est ce sur quoi nous travaillons.
Références :
Ferretti, P., Pasolli, E., Tett, A., Asnicar, F., Gorfer, V., Fedi, S., Armanini, F., Truong, D.T., Manara, S., Zolfo, M., et al.: Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell host & microbe 24(1), 133–145 (2018)
Ricci, L., Heidrich, V., Punčochář, M. et al. Baby-to-baby strain transmission shapes the developing gut microbiome. Nature 651, 191–200 (2026).
Cette étude a été soutenue par des financements ANR : MetaGenoPolis ANR-11-DPBS-0001 et PEPR1071 SAMS PREANALYTICS ANR-24-PESA-0004.
POUR ALLER PLUS LOIN
CroCoDeEL: accurate control-free detection of cross-sample contamination in metagenomic data Nature Communications 2026
Abstract
Metagenomic sequencing provides insights into microbial communities, but it can be compromised by technical biases, including cross-sample contamination. This phenomenon arises when microbial content is inadvertently exchanged among concurrently processed samples, distorting microbial profiles and compromising the reliability of metagenomic data and downstream analyses. Existing detection methods rely on negative controls, which are insufficiently used and do not detect cross-contamination within non-control samples. Meanwhile, strain-level bioinformatics approaches do not distinguish contamination from natural strain sharing and lack sensitivity. To fill this gap, we introduce CroCoDeEL, a decision-support tool for detecting and quantifying cross-sample contamination. Leveraging linear modeling and a pre-trained supervised model, CroCoDeEL identifies specific contamination patterns in species abundance profiles. It requires no negative controls or prior knowledge of sample processing positions, offering improved accuracy and versatility. Benchmarks across three public datasets demonstrate that CroCoDeEL can detect contaminated samples and identify their contamination sources, even at low rates (<0.1%), provided sufficient sequencing depth. Application of CroCoDeEL to several existing studies reveals previously undetected contamination.
Lindsay Goulet, Florian Plaza Oñate, Alexandre Famechon, Benoît Quinquis, Eugeni Belda, Edi Prifti, Emmanuelle Le Chatelier & Guillaume Gautreau. CroCoDeEL: accurate control-free detection of cross-sample contamination in metagenomic data. Nat Commun (2026).
CroCoDeEL
Interface d'interprétation des résultatsDésormais librement accessible à la communauté scientifique internationale, CroCoDeEL constitue un dispositif de contrôle qualité simple pour renforcer la fiabilité des données métagénomiques et propose une interface web. Déjà adopté par plusieurs centres de recherche, il est mobilisé dans des projets d'envergure portés par l'INRAE, tels que French Gut, qui vise à cartographier le microbiote de 100 000 participants.