Food, Global Health Reading time 6 min
CroCoDeEL: a powerful tool for detecting cross-contamination between samples
CroCoDeEL is a powerful decision-support tool designed to detect and quantify cross-contamination between samples during metagenomic analysis. It utilises linear modelling and a pre-trained supervised classifier to identify specific contamination patterns in species abundance profiles.
Published on 13 July 2026
Metagenomic sequencing enables the high-throughput characterisation of complex microbial communities such as the human gut microbiota. However, this approach remains susceptible to a major technical bias: cross-contamination, which is the accidental transfer of DNA from one sample to another during laboratory preparation stages.
If undetected, this contamination leads to microorganisms being attributed to a sample that are in fact absent, thereby compromising the validity of the results.
CroCoDeEL:
Detecting and Quantifying
Contamination
To address this, researchers from INRAE (the MetaGenoPolis and MaIAGE units) and IRD have developed CroCoDeEL (CroCoDeEL is freely available on GitHub), a software designed to detect and quantify such contamination.
The tool relies on the analysis of taxonomic abundance profiles to identify signatures of species transfer between samples, which indicate contamination. Using approaches that combine statistical modelling and machine learning, CroCoDeEL enables the level of contamination to be estimated (even at very low rates) and the source sample to be identified.
Published in Nature Communications in May 2026 under the title: "CroCoDeEL: accurate control-free detection of cross-sample contamination in metagenomic data", the associated study reveals critical instances of contamination that had gone unnoticed in several highly cited landmark studies: certain mother-to-child microbial transfers described in the literature may therefore be technical artefacts rather than proven biological transmissions, calling for a reassessment of previous findings.
Interview with one of the study’s authors: Guillaume Gautreau, a researcher at the INRAE MaIAGE unit.
Why did you want to automate the detection of cross-contamination between samples during high-throughput sequencing?
High-throughput sequencing generates a vast amount of data from a vast number of samples. Contamination can be detected manually, but this is tedious and a Herculean task. Given the sheer volume of samples analysed in high-throughput sequencing, this quickly becomes unmanageable, and it is always possible to miss contamination. For example, we are working on the French Gut project, in which 100,000 samples will be analysed. As the number of possible contamination combinations increases quadratically, it becomes impossible to analyse such a large number of processed samples without a dedicated bioinformatics method.
Furthermore, existing detection methods based on negative controls are underutilised and do not always enable the detection of cross-contamination in non-control samples. At the same time, bioinformatic approaches at the strain level do not always distinguish between genuine contamination and natural strain sharing, and they lack sensitivity.
It is for all these reasons that the automation of contamination detection has quickly become avoidable.
How can cross-contamination between DNA samples be explained?
Contamination can occur in various ways during the handling of samples. Care must be taken when placing samples on plates for analysis, particularly with samples containing low biomass (e.g. meconium), and when carrying out DNA extractions, especially where these are automated, as small droplets may be sprayed and contaminate neighbouring samples. There are many robots in use now, but robots too can cause contamination.
On a more anecdotal note, there are also instances of bioinformatics ‘contamination’. The data produced during high-throughput sequencing represents large volumes, and it can sometimes happen that files are mixed up without anyone realising because automated processes are in place. This mixing is usually detected, but it can waste time by skewing the results in the same way as biological contamination, leading to what is known as in silico contamination.
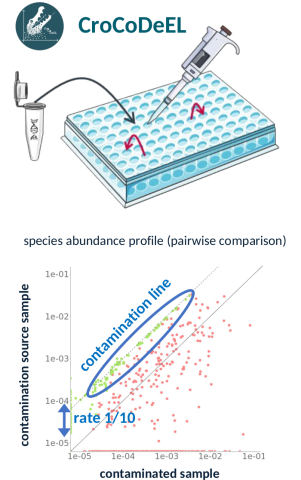
Figure: How does CroCoDeEL detect contamination? When handling dozens of samples on a plate, a small amount of material can be transferred from one well to another. This leaves a signature in the data when two samples are compared. The species introduced by the contamination – which were absent from the sample before it occurred – then align along a single diagonal, the ‘contamination line’. Its position indicates the extent of the contamination: here, 10 per cent of the species content in the contaminated sample originates from its source of contamination.
In your article published in Nature Communications, you detail analyses run with CroCoDeEL on already-published studies, can you tell us more?
Yes, we have applied the CroCoDeEL software to a significant number of studies, and we realised that there was contamination in almost all of them; it is therefore a widespread phenomenon. These contaminations are generally minor, but they reduce the quality of the results, and this could potentially be more expensive because more samples will be needed to obtain a signal free from the background noise caused by the contaminations.
Among these studies, there are two – which we discuss in detail in the article – that reveal very significant contamination or contamination affecting rather critical aspects.
The first study focuses on twins in the United Kingdom. It is a widely cited study (TwinsUK); using CroCoDeEL, we found that 202 out of 1,004 samples were ‘hyper’ contaminated. It is clear that this has a significant impact on the results, as these contaminated samples exhibit an aberrant species richness. We have therefore demonstrated that this problem cannot be ignored. All analyses based on TwinsUK are likely to be biased and will need to be reconsidered.
But the implications extend beyond the TwinsUK study alone. We also applied the method to a study comparing the microbiomes of mothers and their infants (Ferretti et al., 2018). Out of 182 samples, CroCoDeEL identified 48 as contaminated. In our paper, we show that certain species described as the baby’s initial transient colonisers appear to lie on the ‘contamination line’, which is why we believe that some of the conclusions of this study are biased. The study observed high diversity in infants on the first day. Once the contaminated samples were removed, this discrepancy disappeared. Part of what was taken to be a biological signal therefore probably stems from contamination. As a sign that the issue is being taken seriously, in a recent article in *Nature* on microbiota transfers between babies in nurseries, the authors used CroCoDeEL to exclude potentially contaminated samples (Ricci et al., 2026).
CroCoDeEL is now freely available to the entire scientific community, and you have developed an interface for interpreting the results – could you tell us more about it?
The interpretation interface (CroCoDeEL-interpreter) allows you to explore the results visually, directly in your browser. It features graphs comparing samples, and you can integrate the plate layout and contextual data. The visualisations allow contamination trends to be identified at a glance. The idea is that even someone who is not an expert can understand why a particular sample has been flagged and judge for themselves whether the contamination is genuine and whether it is high enough to exclude a sample (the researcher may sometimes prefer to retain a sample if the contamination is minimal). The interface also generates automatic illustrated reports, which compile suspected cases and their graphs: this makes it easy to document the quality control of a dataset, or to share it with colleagues.
CroCoDeEL predictions can be run directly from this interface, without installing anything. A laboratory that is unable to deploy bioinformatics software simply needs to open the page to analyse its data. And as everything takes place in the browser, on the user’s own computer, the data is never sent to a server – an important consideration when working with confidential data.
What are the next steps for CroCoDeEL?
Two projects. The first is to systematise the recording of contamination detected by CroCoDeEL in published studies and to make this information accessible. A few studies are already available in the ‘Datasets’ tab of CroCoDeEL-interpreter, but we wish to go further. This would mean that a researcher reusing this data would know straight away which samples are suspect.
The second, more ambitious project is decontamination. At present, a sample that is too heavily contaminated is simply discarded. However, this means we are discarding data that was costly to produce. We are exploring the design of an in silico method capable of removing the contamination signal whilst preserving what truly belongs to the sample. This is where the difficulty lies: a species introduced through contamination could also be naturally present. Cleaning the data without erasing that signal is what we are working on.
References:
Ferretti, P., Pasolli, E., Tett, A., Asnicar, F., Gorfer, V., Fedi, S., Armanini, F., Truong, D.T., Manara, S., Zolfo, M., et al.: Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell host & microbe 24(1), 133–145 (2018)
Ricci, L., Heidrich, V., Punčochář, M. et al. Baby-to-baby strain transmission shapes the developing gut microbiome. Nature 651, 191–200 (2026).
This work was funded by the MetaGenoPolis grant ANR-11-DPBS-0001 and the PEPR1071 SAMS PREANALYTICS grant ANR-24-PESA-0004.
Notice regarding INRAE’s policy on the use of AI : This translation was carried out helping DeepL translator under human control and revised by the authors.
Find out more
CroCoDeEL: accurate control-free detection of cross-sample contamination in metagenomic data Nature Communications 2026
Abstract
Metagenomic sequencing provides insights into microbial communities, but it can be compromised by technical biases, including cross-sample contamination. This phenomenon arises when microbial content is inadvertently exchanged among concurrently processed samples, distorting microbial profiles and compromising the reliability of metagenomic data and downstream analyses. Existing detection methods rely on negative controls, which are insufficiently used and do not detect cross-contamination within non-control samples. Meanwhile, strain-level bioinformatics approaches do not distinguish contamination from natural strain sharing and lack sensitivity. To fill this gap, we introduce CroCoDeEL, a decision-support tool for detecting and quantifying cross-sample contamination. Leveraging linear modeling and a pre-trained supervised model, CroCoDeEL identifies specific contamination patterns in species abundance profiles. It requires no negative controls or prior knowledge of sample processing positions, offering improved accuracy and versatility. Benchmarks across three public datasets demonstrate that CroCoDeEL can detect contaminated samples and identify their contamination sources, even at low rates (<0.1%), provided sufficient sequencing depth. Application of CroCoDeEL to several existing studies reveals previously undetected contamination.
Lindsay Goulet, Florian Plaza Oñate, Alexandre Famechon, Benoît Quinquis, Eugeni Belda, Edi Prifti, Emmanuelle Le Chatelier & Guillaume Gautreau. CroCoDeEL: accurate control-free detection of cross-sample contamination in metagenomic data. Nat Commun (2026).
CroCoDeEL interpreter
Results interpretation interfaceNow freely available to the international scientific community, CroCoDeEL is a simple quality control tool designed to enhance the reliability of metagenomic data and features a web interface. Already adopted by several research centres, it is being used in large-scale projects led by INRAE, such as French Gut, which aims to map the microbiota of 100,000 participants.