Biodiversité Temps de lecture 5 min
Un logiciel qui connecte des bases de données pour étudier les réseaux de régulation des voies biologiques
Publié le 08 février 2022

Les phénotypes animaux sont déterminés par des caractères complexes impliquant un grand nombre de voies biologiques. L’orientation des phénotypes est possible en favorisant une voie biologique plutôt qu’une autre et/ou en effectuant une sélection génétique sur un caractère particulier ; pour cela, il est important de définir les éléments clés susceptibles de jouer un rôle pivot dans le contrôle des réponses aux contraintes génétiques ou environnementales, et d’identifier les relations entre les entités biologiques à différents niveaux d’organisation du vivant.
Il existe des sources de données ouvertes et en ligne décrivant les entités biologiques (acides nucléiques, protéines, complexes, métabolites...) pour différents organismes, sachant que pour la plupart, elles concernent l’espèce humaine. Pour pouvoir interroger une ou plusieurs de ces bases de données et en combiner les informations, il est nécessaire de disposer d’une solution globale d’homogénéisation des informations et d’une simplification de leurs relations aux différents niveaux d’organisation du vivant.
Les chercheurs de l’UMR PEGASE ont développé un logiciel open source (baptisé PAX2GRAPHML) permettant d’étudier des réseaux biologiques sous forme de grands graphes de réactions. Les données de 17 sources internationales ouvertes et portant sur des entités biologiques de mammifères ont été ainsi prétraitées et sont mises à disposition sous la forme de fichiers standardisés. Un travail important de modélisation a été réalisé pour décrire de manière homogène les entités biologiques (acides nucléiques, protéines, complexes, métabolites) selon le standard BIOPAX*, et pour représenter leurs relations de manière simplifiée permettant de relier les niveaux de régulation, de signalisation et du métabolisme.
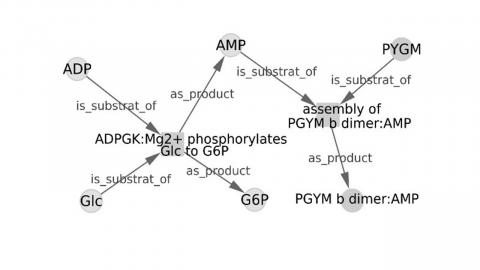
L’outil permet ainsi d’interroger une ou plusieurs de ces sources de données et de proposer une représentation graphique des relations entre entités sous forme de graphes de réactions à large échelle. Il constitue une contribution importante pour rendre les données interopérables. Il permet ainsi de produire des connaissances pour mieux comprendre les réponses cellulaires et leur adaptation aux contraintes génétiques ou environnementales.
Les applications de ce logiciel sont nombreuses et consisteront en particulier à trouver des régulateurs importants des phénotypes qui pourront alors être ciblés par des stratégies génétiques ou nutritionnelles adaptées, d’en prévoir les effets connexes ou bien de favoriser une voie métabolique particulière.
Ces travaux ont été conduits dans le cadre d’une collaboration entre l’UMR PEGASE/INRAE et l’IRISA/INRIA, illustrant la plus-value d’un dialogue entre biologistes, informaticiens et mathématiciens. Le logiciel va être amélioré grâce à un interfaçage avec des outils d’analyses topologiques de graphes et de visualisation plus avancée.
* Biological Pathway Exchange (BioPAX) est un langage standard qui vise à permettre l'intégration, l'échange, la visualisation et l'analyse des données sur les voies biologiques.
Disponible sur https://pax2graphml.genouest.org.
François Moreews, Hugo Simon, Anne Siegel, Florence Gondret, Emmanuelle Becker, PAX2GRAPHML: a python library for large-scale regulation network analysis using BioPAX, 2021; Bioinformatics, 37 (24): 4889-4891. https://doi.org/10.1093/bioinformatics/btab441